

搜索



新闻中心
NEWS
资讯分类
侧边栏
发布时间:2020-03-11 00:00:00


新闻详情
一文看懂FDA cGMP 483解析
- 分类:行业资讯
- 作者:巍信
- 来源:东富龙系统部
- 发布时间:2016-11-14 12:00
- 访问量:
【概要描述】摘要FDA483报告,也称现场检查报告,是美国FDA检查人员根据美国现行法律法规,对企业进行现场检查,对不符合cGMP的地方进行总结并发给企业。企业需立即针对不符合项进行整改,并在收到483报告后15个工作日内向FDA相关部门回复483中不符合项的整改情况。若FDA认定企业的答复妥当,并符合现行法律法规的要求,那么企业就避免收到FDA警告信。FDA每个财政年

一文看懂FDA cGMP 483解析
【概要描述】摘要FDA483报告,也称现场检查报告,是美国FDA检查人员根据美国现行法律法规,对企业进行现场检查,对不符合cGMP的地方进行总结并发给企业。企业需立即针对不符合项进行整改,并在收到483报告后15个工作日内向FDA相关部门回复483中不符合项的整改情况。若FDA认定企业的答复妥当,并符合现行法律法规的要求,那么企业就避免收到FDA警告信。FDA每个财政年
- 分类:行业资讯
- 作者:巍信
- 来源:东富龙系统部
- 发布时间:2016-11-14 12:00
- 访问量:
详情
摘要
FDA483报告,也称现场检查报告,是美国FDA检查人员根据美国现行法律法规,对企业进行现场检查,对不符合cGMP的地方进行总结并发给企业。企业需立即针对不符合项进行整改,并在收到483报告后15个工作日内向FDA相关部门回复483中不符合项的整改情况。若FDA认定企业的答复妥当,并符合现行法律法规的要求,那么企业就避免收到FDA警告信。FDA每个财政年都会统计、总结并发布483表格,其在官网上公布。本文通过对FDA官网发布的2010年~2015年483数据进行简要分析,并着重对FDA cGMP 483进行说明和简要分析,旨在为制药企业完善内部管理体系提供一定的借鉴作用。
1数据概览
1、1483数据概况
FDA,中文全称为食品药品监督管理局,其监管的范围覆盖了食品、各种类型的药品、医疗器械等行业,表1列出了2010~2015财政年各个领域的检查总结情况。
表1 2010~2015财政年发布的483份数
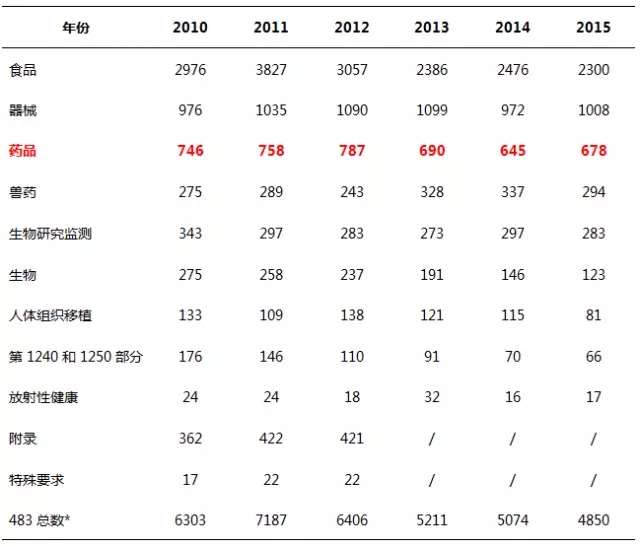
注:*以上表格并不代表财政年发布的所有483份数,因为部分483为手写版,未包含在该表格中。
纵观各个领域的483数据,食品483比重最大,约占总数的一半。药品483比重从2010至2015财政年分别为11.9%、10.5%、12.3%、13.2%、12.7%、14.0%。表2为2010~2015财政年药品483数量情况。
表2 2010~2015财政年药品483数量
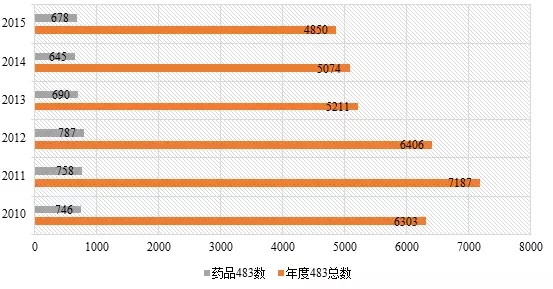
2、药品483概况
药品483涉及FDA颁发的联邦法规第21章药品相关法规和法律,包括成品药良好生产管理规范、PET药品良好生产管理规范、新药、联邦食品药品化妆品法律等。表3列出了2010~2015财政年各法规出现不符合项的频次。
表3 2010~2015财政年各法规出现不符合项的频次
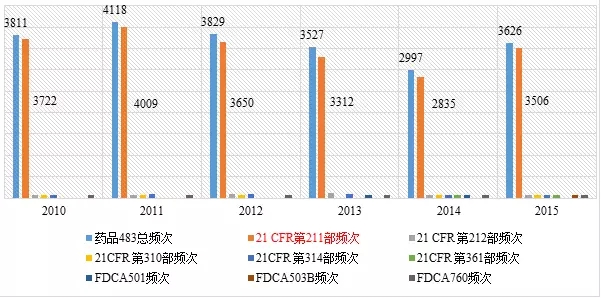
其中,21 CFR第211部是成品药良好生产管理规范(现行版本生效日期为2014年4月1日),21 CFR第212部是PET药品良好生产管理规范(现行版本生效日期为2014年4月1日),21 CFR第310部是新药(现行版本生效日期为2014年4月1日),21CFR第314部是新药上市FDA批准申请(现行版本生效日期为2014年4月1日);21CFR第361部为认为安全、有效、未贴假商标的处方药:调查用药(现行版本生效日期为2014年4月1日);FDCA501:联邦食品、药品和化妆品法501;FDCA503:联邦食品、药品和化妆品法503;FDCA760:联邦食品、药品和化妆品法760。
从表3的数据可知,21CFR第211部收到483报告的频次最多,分别占药品483总频次97.7%、97.4%、95.3%、93.9%、94.6%、96.7%。而21CFR第211部是美国FDA对药品生产的基本要求,是FDA检查人员检查药企遵循的重要准则。
3、FDA cGMP 483数据概况
FDA cGMP即21CFR第211部。表4按照六大体系展示不符合项的分布情况。
表4药品cGMP483分布(按照六大体系划分)
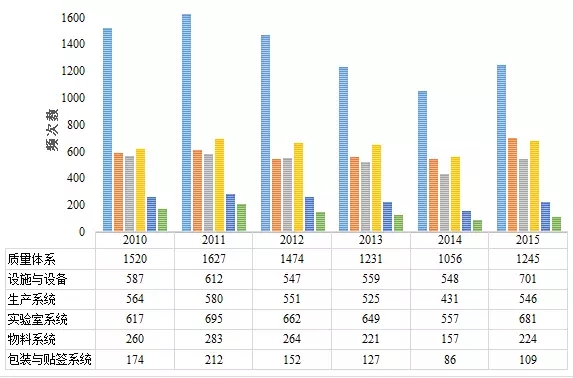
表4数据显示,在六大体系中,质量体系出现不符合项的次数最多,大约占cGMP不符合项总数2/5;实验室检验系统次之,比重大约为cGMP不符合项总数1/6;厂房设施与设备系统和生产系统基本并列第三,约占cGMP不符合项总数1/7。
2六大体系483情况
1、质量体系483情况
根据表4的数据计算,2010~2015财政年质量体系出现不符合项占cGMP不符合项总数分别为40.8%、40.6%、40.4%、37.2%、37.2%、35.5%,逐年减少。表5汇总了质量体系出现缺陷的部分情况。
表5 质量体系不符合项部分汇总
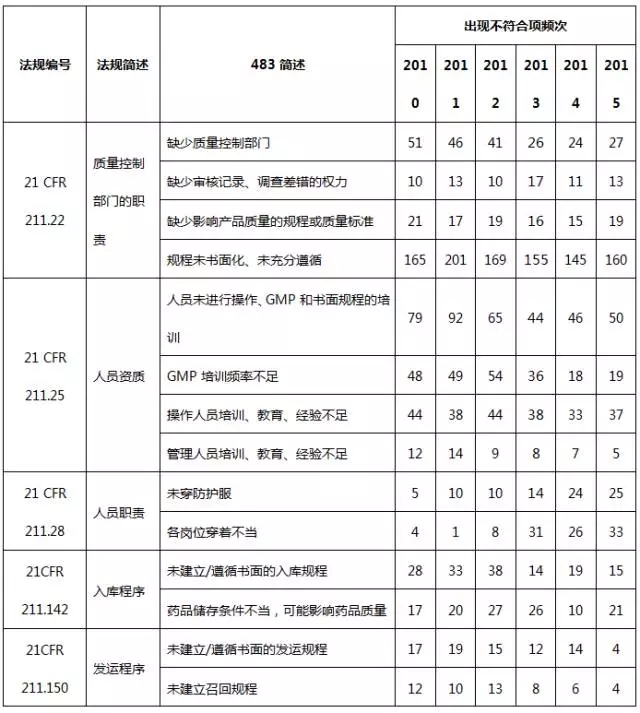
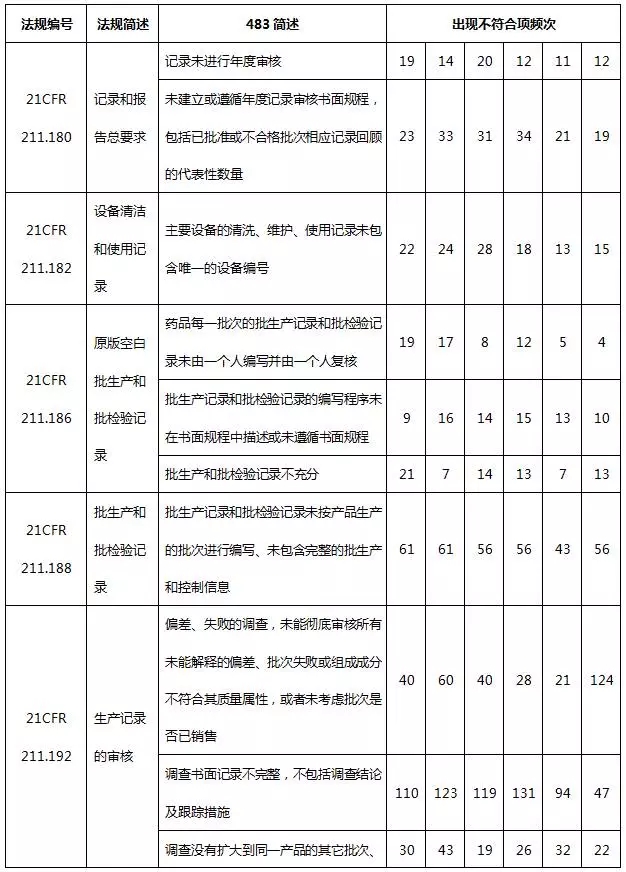
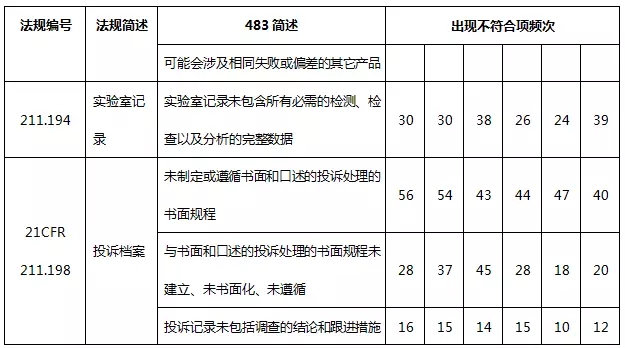
质量管理体系出现很多不合规的现象,主要表现于质量体系不健全,特别是文件体系,缺乏书面的质量管理规程或没有充分遵守已经制订的规程,包括QC/QA管理规程、员工培训规程、入库规程、发运规程以及各种记录。另外,人员教育、培训及实际经验缺乏也是常见的缺陷项。
制药企业的质量体系水平在一定程度上体现了企业的质量文化水平,实际上质量体系缺陷是一个国际性问题,其不仅出现在FDA 483中,在EudraGMDP不符合项报告中也很常见。制药企业应该提高质量文化的建设,建立完善的质量体系,并定期评估质量体系运行的有效性。
2、实验室检验系统483
2010~2015财政年实验室检验系统不符合项出现概率分别占16.6%、17.3%、18.1%、19.6%、19.6%、19.4%。以下就实验室检验系统不符合项进行部分汇总。
表6 实验室检验系统不符合项部分汇总
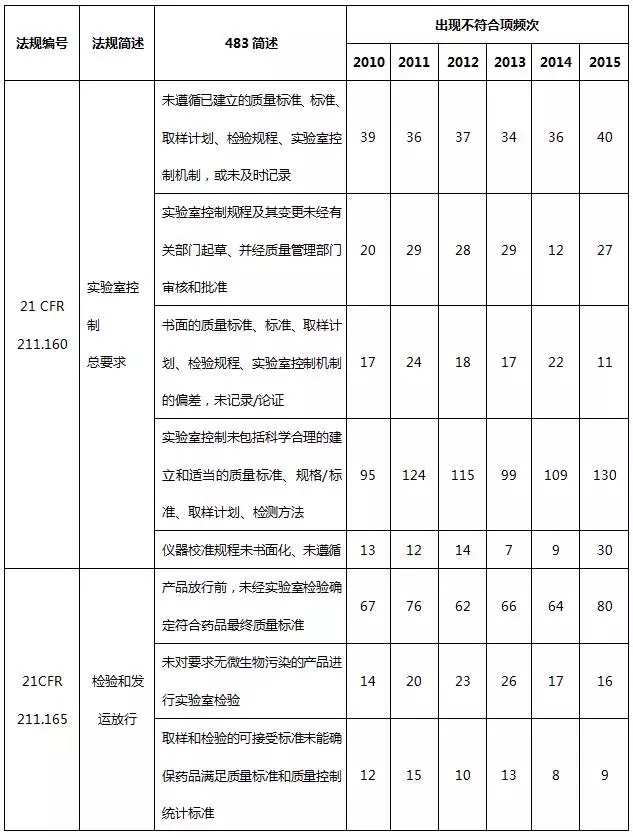
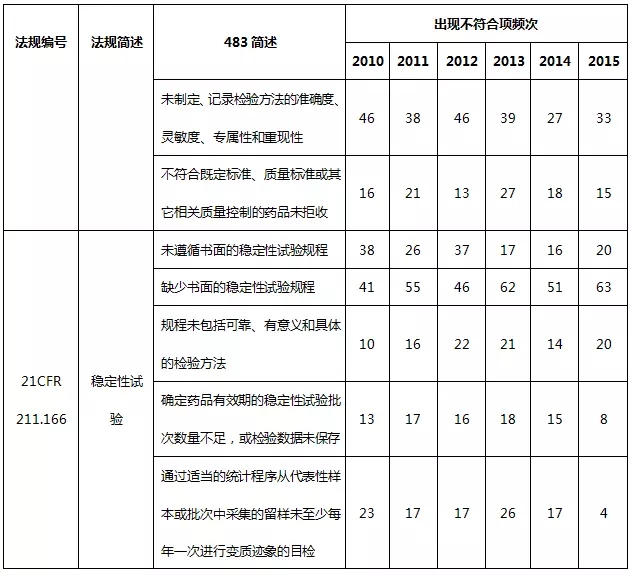
实验室检验系统缺陷主要体现在质量标准、取样计划、检验规程、实验室控制机制相关规程未建立、未充分遵循以及未及时记录,特别是实验室控制规程,包括实验室数据完整性、真实性、准确性,是频发的不合规问题。当下制药行业再次热议数据完整性(CFDA已正式更名为数据可靠性)这一话题,不管是手工记录还是电子记录,均应以诚信为基石。制药企业可通过完善数据与记录管理的技术基础、完善记录管理程序、完善记录文件、建立计算机化系统管理系统或高素质的员工培训来确保数据的完整性。
3、厂房设施与设备系统483情况
2010~2015财政年厂房设施与设备系统不符合项出现概率分别占15.8%、15.3%、15.0%、16.9%、19.3%、20.0%。表7就厂房设施与设备系统不符合项进行部分汇总。
表7 厂房设施与设备系统不符合项部分汇总
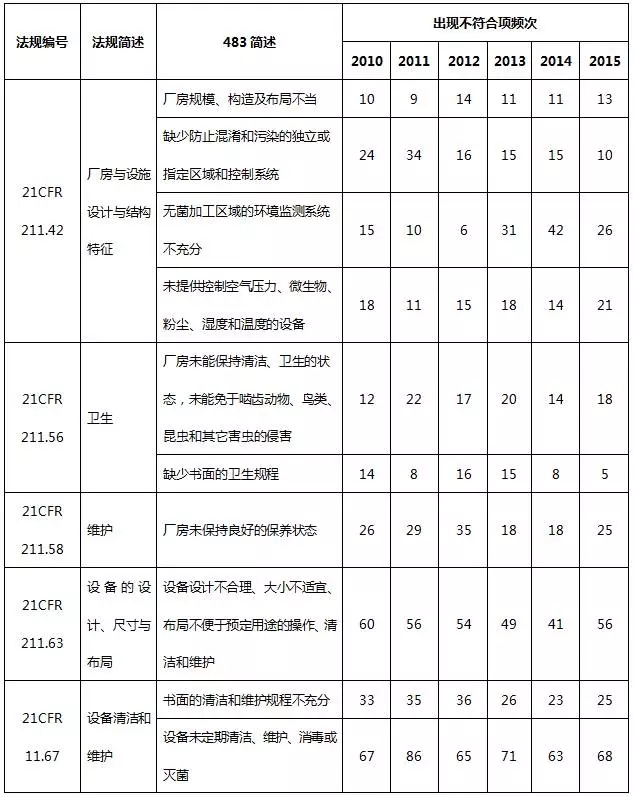
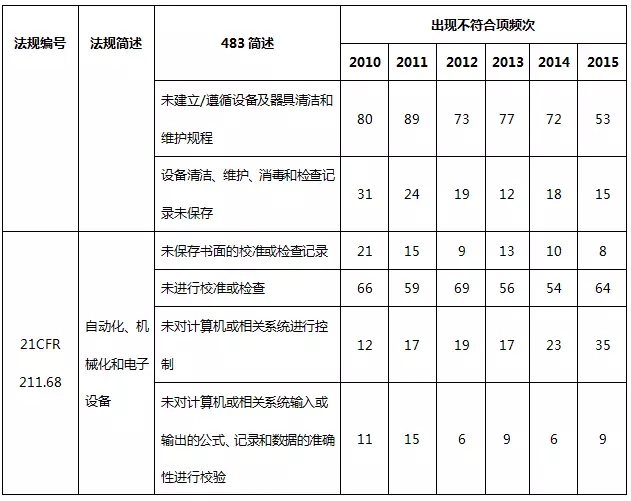
在厂房设施与设备系统中,预防交叉污染、混淆和差错不充分,设计、布局不合理,不利于操作、清洁和维护是主要的缺陷项目。自动化设备或仪器没有进行准确性确认、校准或控制,也时常被发现。
交叉污染是GMP的三大核心之一,制药企业应详细评估车间布局的可行性,并进行严格的验证活动,避免挑战法规的要求。自动化设备则应按照相关法规进行验证,如CFDA最新颁布的GMP附录,计算机化系统。
4、生产系统483情况
2010~2015财政年生产系统不符合项出现概率分别占15.2%、14.5%、15.1%、15.9%、15.2%、15.6%。以下就生产系统不符合项进行部分列表。
表8 生产系统不符合项部分汇总
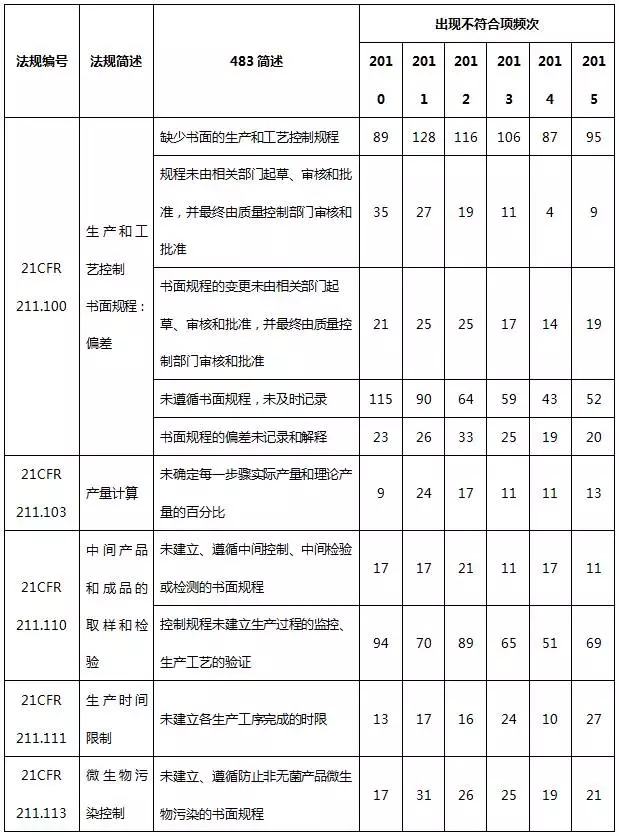
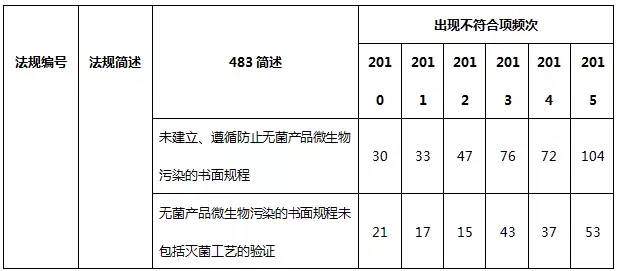
生产系统常见的不合规项主要是缺少生产和工艺控制、微生物污染控制书面规程、未遵循书面规程、未及时记录,未对每一步骤进行产量计算以及未进行灭菌工艺验证。值得一提的是,国内制药企业通常会忽略除热原工艺验证,这在FDA认证中将构为严重缺陷。工艺验证按照2011版FDA行业指南——工艺验证的一般原则和规范提供的方法进行,其应贯穿药品的整个生命周期,包括药物研发阶段,一直到产品退市。
5、物料系统483情况
2010~2015财政年物料系统不符合项出现概率分别占7.0%、7.1%、7.2%、6.7%、5.5%、6.4%。以下就物料系统不符合项进行部分列表。
表9 物料系统不符合项部分汇总
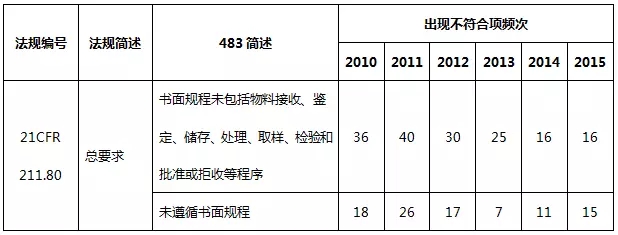
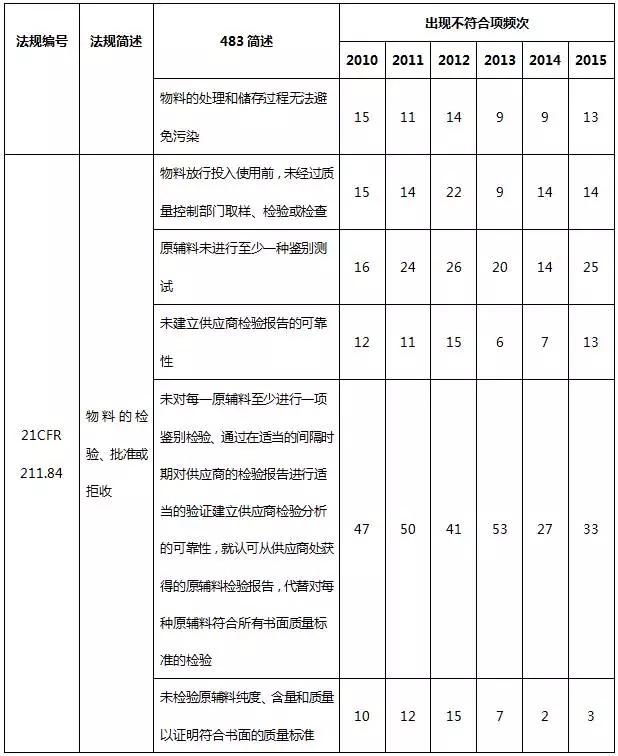
物料系统出现违规项目主要是书面规程不足、物料检验过分依赖供应商检验报告而未进行检验。从严格意义上来说,表9列举的缺陷项属于质量体系的一部分,这就需要制药企业提高质量的意识,从各个环节严格把控药品生产的质量。
6、包装和贴签系统483情况
2010~2015财政年包装和贴签系统不符合项出现概率分别占4.7%、5.3%、4.2%、3.8%、3.0%、3.1%。表10汇总了包装和贴签系统出现不符合项的部分情况。
表10包装和贴签系统不符合项部分汇总
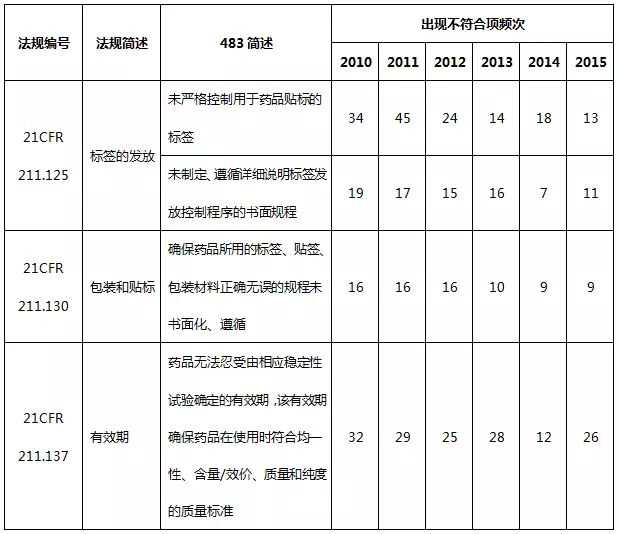
包装和贴签系统依旧存在规程未书面化或没有充分遵循的情况,此外有效期的确定不够科学。同样地,包装和贴签系统的缺陷项在一定程度上也体现为质量管理体系的不足。
3结束语
随着国内制药企业水平的不断提高,不少企业欲向欧美市场进军,通过FDA cGMP是最低门槛,本文信息仅作为制药企业对照现有的质量保证体系是否存在违反FDAcGMP的参考。特别是在规程书面化、完整化以及充分遵循化方面,不管是在质量体系还是包装贴签体系,对出现的违规现象应该引起广大制药企业的深思和警醒。
扫二维码用手机看
上一个:
无
下一个:
CFDA放大招,自查之后将铁血飞检!
上一个:
无
下一个:
CFDA放大招,自查之后将铁血飞检!
页面版权所有 湖北亿诺瑞生物制药有限公司 地 址:湖北省黄梅县小池镇沿江路108号 证书编号:(鄂)-非经营性-2018-0070